Maurizio Carbonari*, Marina Cibati*, Anna Maria
Pesce*, Roberto Gradini, Andrea Modesti & Massimo Fiorilli*.
*Dipartimenti di Medicina Clinica e di Medicina Sperimentale e Patologia, Università di Roma "La Sapienza".
This study was supported by grants from the Italian Ministry of Health, VIII Progetto AIDS, and from the Istituto Pasteur-Fondazione Cenci Bolognetti (to M.F). M.Ci. was supported by a postdoctoral fellowship from the Italian Ministry of Health.
Corresponding author:
Maurizio Carbonari
Department of Clinical Medicine,
University of Rome 'La Sapienza',
Viale dell'Università 37,
00185 Rome, Italy.
Fax: 06-4463877
Phone: 06-49972034
Flow cytometry is one of the most powerful and specific methods for the integrated study of the molecular and morphological events occurring during cell death and cell proliferation, and many methods have been described for investigating these phenomena. Ideally, methods should allow the contemporaneous determination of morphology, phenotype, intracellular protein expression, and status of chromatin and of DNA; this would make possible to assess the type and mechanism of cell death and the cell cycle position at the single cell level. Toward this end, we have developed a simple procedure of fixation/permeabilisation that permits quantifying DNA content, identifying membrane (and possibly intracellular) antigens, and satisfactorily preserving the optical properties of the cells (i.e. their light scatter characteristics). This technique works well with a variety of cytotypes, including human whole peripheral blood leukocytes which, in our hands, are hardly amenable to such analysis by other methods. Preliminary results suggest that, under certain conditions, paradoxical modifications of the apparent DNA content (i.e. hyperdiploidy) may be related to early necrobiological changes.
Key words: differential light scatter, ploidy, phenotype, cell cycle, apoptosis.
Cell death, and particularly apoptosis, has recently raised a wealth of interest among biomedical researchers and businessmen (1,2). Even the taxonomy of cell death is in evolution, and neologisms such as "necrobiosis" and "oncosis" have been proposed to indicate, respectively, the whole biological changes which predispose and accompany cell death (1) and those forms of accidental cell death usually referred to as necrosis (3).
It is likely that, in addition to the extremes of programmed death by apoptosis and of accidental death by necrosis, other forms of cell death exist (4). In some cases it appear to exist admixtures of apoptosis and necrosis, made evident by the coexistence of morphological and biochemical features corresponding to these processes (5,6). It is lilkely that these unusual forms of cell death will be unveiled by more deeply investigating in vivo models, where the complex environmental influences might smoothen the polarisation between fully organized (apoptotic) and fully disorganised (necrotic) forms cell death seen in in vitro models.
In this setting, flow cytometry offers an invaluable tool of investigation (1). Last generation flow cytometers, with improved stability of fluidics and efficiency of optics as well as with expanded detection channels, allow tremendous levels of signal resolution of both differential light scatters (DLS) and of fluorescences. These instruments permit to resolve minute differences in cell size and structure and in the uptake of fluorescent probes, and are therefore suitable for detailed multiparameter analyses of DNA content and morphology and antigenic makeup of the cells. Unfortunately, current methods of fixation/permeabilisation for the measurement of nuclear DNA profoundly alter the light scatter characteristics of the cells. On the other hand, fixation procedures aimed at preserving the intrinsic optical characteristics of the cells do not warrant linearity between the amount of nuclear DNA and the height/width of signals by fluorescent intercalating dyes (7). Finally, even those protocols which sufficiently preserve DLS and antigenic properties of the cells, yet permitting DNA quantification (7), may fail when used with cell populations which include cytotypes rich of enzymes such as polymorphonuclear (PMN) leukocytes.
To address these problems, we tested whether the simultaneous use of permeabilising/fixing (e.g. ethanol) and of fixing/permeabilizing (e.g. formaldehyde) agents could result in reliable DNA quantification while preserving cell morphology and phenotype, and whether it could be applied to "problematic" cell types.
Whole human peripheral blood leucocytes were obtained from EDTA-treated venous blood after the lysis of erythocytes with the Ortho Lysing Reagent® (Ortho, Raritan, NJ); human peripheral blood mononuclear cells were isolated by density gradient centrifugation (Lymphoprep, Nyeegard, Oslo, Norway). Single cell suspensions from rat thymuses were obtained as previously described (6).
Cell Staining and Flow Cytometry.
Cells were stained with monoclonal antibodies (MAbs) to surface antigens and with DNA dyes according to the following procedure, which was established following a series of preliminary experiments.
Cells (between 5x105 and 2x106 in 100 ml of PBS containing 2% fetal calf serum) were stained with saturating concentrations of fluochrome-conjugated Mabs to leukocyte surface antigens (alll from Becton-Dickinson) at 4°C for 30 minutes. Cells were then washed by the addition of 40 volumes (4 ml) of Ortho Lysing Reagent® for 15 min. at room t°, followed by centrifugation for 7 min. at 250 g at 20°C. Pelleted cells were gently resuspended in 1 ml of a solution (indicated as A.2 solution) containing 5% 1,2-propanediol, 0.75% formaldehyde, 30% ethanol, and 150 mM natrium citrate, and incubated for 30 min. at room temperature. After incubation, cells were pelleted at 800 g for 5 min. at 20°C, and further washed with 1 ml of 150 mM natrium citrate. DNA staining was done by incubating cells for 16 hours at room t° in 1 ml of 150 mM natrium citrate containing 10 mg/ml of 7-amino-actinomycin D (7-AAD) (Sigma, St. Louis, MO). Cells were analysed by flow cytometry without further washings.
In some experiments, analysis of apoptosis was performed either by the procedure of Telford et al. (7) for hypodiploidy, or by the TUNEL assay (8) (Boehringer Mannheim, according to the manufacturer's instructions except for the use of the A.2 solution for cell permeabilisation).
Flow cytometry was done using a FACSCalibur instrument
(Becton Dickinson, San Jose, CA) with argon laser excitation at 488 nm.
Multiparameter analysis of DNA content in mixed cell populations.
We preliminarily examined mixed leukocyte populations containing PMN by a published technique for the analysis of DNA content in in whole cells (7). The light scatter properties of the cells, as well as the stainability of surface antigens, were completely subverted by this treatment (even if extreme care was put in performing all steps according to the Author's indications, ref. 7, particularly controlling temperatures and the rate of addition of hydroalcoholic solution). This fact was seemingly due to the presence of PMN, since peripheral blood mononuclear cell (PBMC) preparations could be adequately stained with MAbs and DNA dyes although the light scatter properties were very poorly preserved (data not shown).
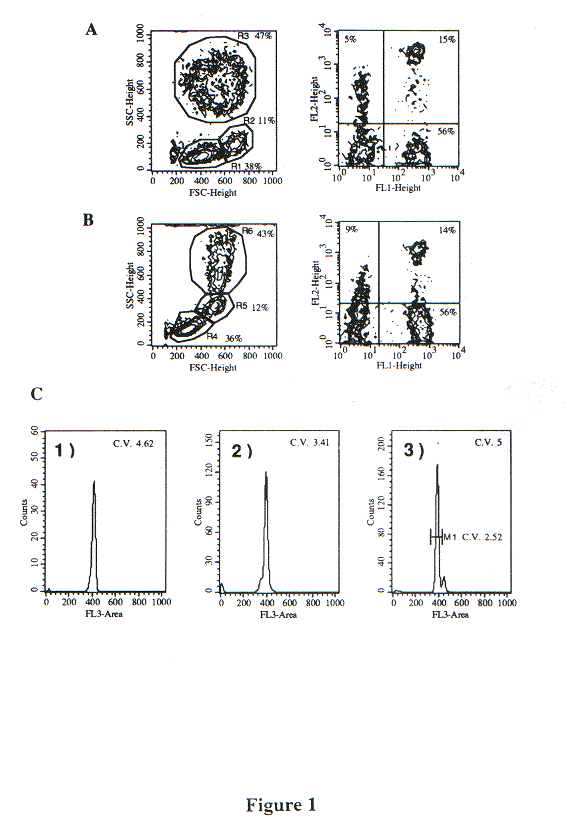
By contrast, fixation with our A.2 solution (see Methods) did not alter significantly the DLS properties of the major leukocyte populations (lymphocytes, monocytes and PMN) (Fig. 1); furthermore, the stainability of surface antigens with MAbs (performed before fixation) was identical to that of unfixed cells. DNA staining with 7-AAD gave histograms with a CV lower than 5%, permitting adequate evaluation of DNA content. It is particularly relevant that the DLS characteristics were sufficiently preserved to permit the differential gating on distinct leukocyte populations.
Measurement of DNA in apoptotic thymocytes: facts or artifacts?
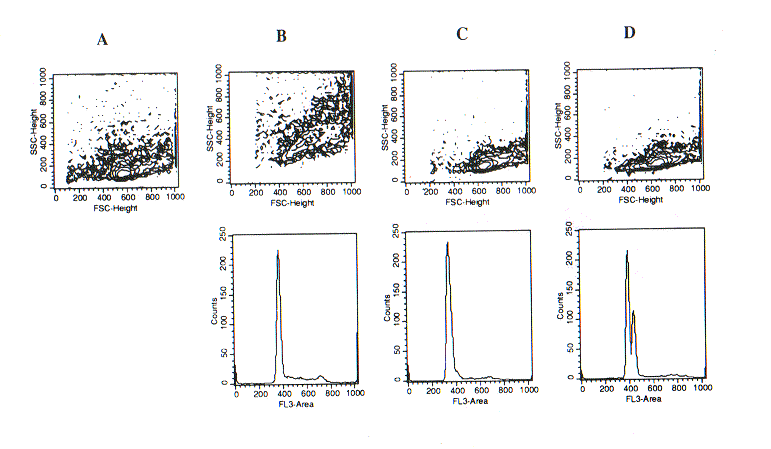
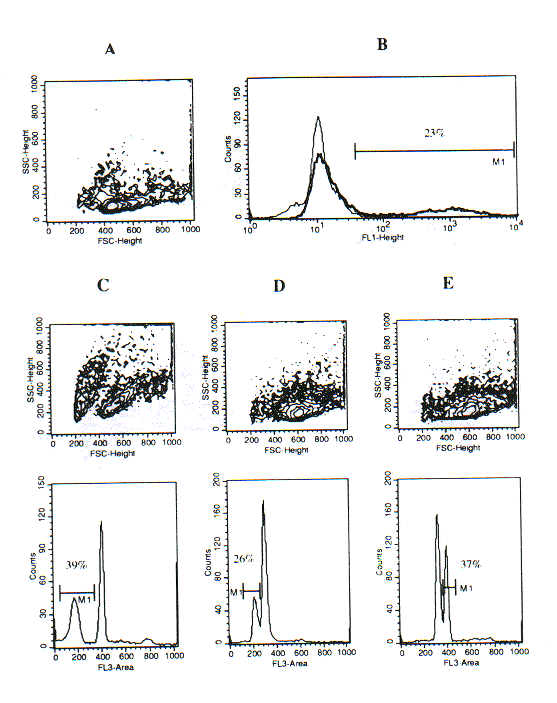
Since our fixation/permeabilisation procedure described above appeared suitable for the accurate measurement of cellular DNA content, we tested whether it could be applied to the detection of apoptotic cells based on their apparent hypodiploidy (1). The results of experiments with rat thymocytes were, indeed, intriguing, since they revealed that subtle differences in sample preparation could dramatically affect the apparent DNA content of apoptotic (or pre-apoptotic) thymocytes. Ethanol permeabilisation of freshly isolated thymocytes according to the procedure of Telford et al. (7) determines an appreciable increment of SSC and FSC, and DNA staining with 7-AAD reveals a major diploid peak and a fraction of cells in the S or M phase of the cell cycle (Fig. 2B). Direct fixation of thymocytes with the A.2 solution yelds well preserved light scatters and a similar DNA profile (Fig. 1C). However, when thymocytes were treated with the Ortho Lysing Solution (as described in Methods) before treatmetn with the A.2 solution the major diploid peak was plit into two components, the minor of which was slightly hyperdiploid (Fig. 2D). The analysis of the width and height of FL3 fluorescence revealed that the bymodal distribution of the FL3-area hystogram depended on different levels of intensity of nuclear fluorescence. This was confirmed by confocal microscopy of the cell samples (data not shown). It was tempting to speculate that the hyperdiploid sub-peak corresponded to apoptotic or pre-apoptotic thymocytes with modified chromatin conformation. To clarify this issue, we combined our procedure with the TUNEL assay for DNA fragmentation (8). Fixation/permeabilisation with the A.2 solution turned out to be fully compatible with tdt activity in the TUNEL assay, and thus this technique could be combined with the analysis of DNA content using 7-AAD staining. As shown in Fig. 3, apoptotic thymocytes, recovered after 24-hour in vitro culture and treated with lysing solution before fixation, had cleaved DNA (FL1-fluorescence) and displayed an apparently hyperdiploid DNA content (FL3 fluorescence), while a fraction of hyperdiploid cells was TUNEL-negative. By contrast, in samples that were not treated with the lysing solution before fixation there was not an hyperdiploid sub-peak, but rather it gecame vident a sub-diploid population whose magnitude was close to that detected by the Telford's assay and to the populaion of TUNEL-positive cells (Fig. 4).
DISCUSSION
The techniques which analyse the cellular DNA content can be divided in two groups: the first one makes use of isolated nuclei, while the second exploits more or less well-preserved whole cells. In the first case, when the assay is aimed at evaluating hypodiploidy as an indicator of apoptosis (9), there is the serious risk of overestimating the number of sub-G0/G1 events. This happens because of the possible presence in the sample of nuclear fragments, single chromosomes, chromosome clumps, cellular debris, contaminating bacteria, etc. This risk is particularly high when the DNA fluorescence is acquired after logarithmic amplification of light impulses (1). The application of linear rather than logarithmic amplification allows to distinguish hypodiploid events from objects with a minimal DNA content. Furthermore, the use of isolated nuclei precludes the evaluation of other cellular parameters.
For these reasons, methods using whole cells are greatly preferable. Rabinovitch et al. (10) were the first to propose the simultaneous analysis of cellular cycle and two-color surface immunofluorescence. In spite of a very good intercompatibility of the fluorochromes (7-AAD, FITC and PE), in our hands this method showed significant limits concerning both the conservation of DLS and the applicability to different cytotypes. More recently, Schmid et al. (11) described a gentle fixation with a polymerising agent followed by a mild non-ionic detergent treatment. This approach fulfills the requirements for a multiparametric analysis. However, the sample is extremely labile unless one strengthens the fixation step, but this may reduce its performance since extensive fixation by cross-linking is not compatible with a linear quantitation of DNA content and with a clear separation between G0/G1 and sub-G0/G1 peaks (7). Accordingly, we were unable to obtain satisfactory nuclear fluorescence histograms with cells (cycling or apoptotic) prefixed with various concentrations of glutaraldehyde, formaldehyde or paraformaldehyde, at different temperatures and incubation times. This was true irrespectively of the permeabilising agent employed (ethanol, triton X-100, tween-20, NP-40). Rather paradoxically, the nuclear fluorescence intensity of apoptotic (x-irradiated) human PBMC stained with 7-AAD after cross-linking fixation was higher than that of normal PBMC. In our experience, the method described by Zarbo et al. (12), and subsequently modified by Garvy et al. (13) and by Telford et al. (7), is the best one for the multiparametric analysis of DNA content. This procedure, when carefully executed, is very efficient with cytotypes like proliferating and/or apoptotic PBMC and cell lines. However, as stated above, this method is unsuitable with samples containing polymorphonuclear leukocytes. This is probably due to poor fixation, as suggested by the ultrastructural changes (our unpublished data) and the cronolability of samples treated in this way.
In an attempt to overcome the above indicated limits of the current fixation/permeabilisation methods, we developed a procedure based on the following considerations. Formaldehyde fixes cells and partially permeabilises their membranes, while ethanol at low concentrations completely permeabilises cells although preserving some of their morphological features (size, absolute and relative refraction indices of surface and internal structures). Thus, we started with a solution containing these components, and empirically modified its composition up to an optimal fixative (maintenance of cell morphology) and permeabilising (CV ?5% on DNA staining with 7-AAD) activity. Among the membrane-stabilising agents tested, 1,2-propanediol showed both the highest capacity to preserve cellular morphology and the least interference on fluorescence emissions. As saline component in the fixation/permeabilisation and in the DNA staining steps, natrium citrate appeared the best in reducing the coefficient of variation and increasing the intensity of nuclear fluorescence. The consequent red fluorescence gain allowed reducing the concentration of 7-AAD, thus making easier to adjust compensations. Maintaining samples at low temperature turned out to be of paramount importance for preserving the morphology (i.e. light scatter properties) of "difficult" samples such as those containing granulocytes. In the case of this type of samples all the other previously described procedures gave, in our hands, quite poor results even when temperatures were rigorously controlled.
Finally, the quite fortuitous addition of a step
of "erythocyte lysis" (using a solution containing ammonium chloride) before
cell fixation by our protocol revealed intriguing effects on DNA stainability.
In fact, although this step generally improved the indices of nuclear fluorescence
histograms, when used with samples containing apoptotic cells resulted
in a hyper-diploid peak which, paradoxically, appeared to correspond to
the apoptotic population, and which was not detectable when the "lysis"
step was omitted. It is possible that this phenomenon depends on changes
of the chromatin conformation of apoptotic (or pre-apoptotic) cells which,
under the action of ammonium chloride, render DNA more accessible to certain
dyes. Thus, although the presently described method of fixation/permeabilisation
may represent a significant evolution toward a fully multiparameter flow
cytometric analysis of the cell cycle, it underscores the complexity of
approaching the evaluation of apoptosis by the measurement of apparent
cell ploidy.
1) Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F: Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death. Cytometry 27:1-20, 1997.
2) Cohen JJ, Al-Rubeai M: Apoptosis-targeted therapies: the 'next big thing' in biotechnology? Trends Biotechnol 13:281-283, 1995.
3) Majno G, Joris I: Apoptosis, oncosis and necrosis: an overview of cell death. Am J Pathol Am. J. Pathol. 146: 3-15,1995.
4) Schwartz LM, Osborne BA: Programmed cell death, apoptosis and killer genes. Immunol Today 14:582, 1993.
5) Pulendran B, Van Driel R, Nossal GJV: Immunologicval tolerance in germinal centres. Immunol Today 18:27-32, 1997.
6) Carbonari M, Pesce AM, Cibati M, Modica A, Dell'Anna L, D'Offizi G, Angelici A, Uccini S, Modesti A, Fiorilli M. Death of bystander cells by a novel pathway involving early mitochondrial damage in HIV-related lymphadenopathy. Blood in press.
7) Telford WG, King LE, Fraker PJ: Rapid quantitation of apoptosis in pure and heterogeneous cell populations using flow cytometry. J Immunol Meth 172:1, 1994.
8) Gavrieli Y, Sherman Y, Ben-Sasson SA: Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119:493, 1992
9) Nicoletti I, Migliorati G, Pagliacci MG, Grignani F, Riccardi C: A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Meth 139:271-279, 1991.
10) Rabinovitch PS, Torres RM, Engel D: Simultaneous cell cycle analysis and two-color surface immunofluorescence using 7-amino-actinomycin D and single laser excitation: applications to study of cell activation and the cell cycle of murine Ly-1 B cells. J Immunol 136: 2769-2775, 1986.
11) Schmid I, Uittenbogaart CH, Giorgi JV: A gentle fixation and permeabilization method for combined cell surface and intracellular staining with improved precision in DNA quantification. Cytometry 12: 279-285, 1991.
12) Zarbo RJ, Visscher DW, Crissman JD: Two-color multiparametric method for flow cytometric DNA analysis of carcinomas using staining for cytokeratin and leukocyte common antigen. Anal Quant Cytol Histol 11: 391-402, 1989.
13) Garvy BA, Telford WJ, King LE, Fraker PJ: Glucocorticoids- and irradiation-induced apoptosis in normal murine bone marrow B lineage lymphocytes as determined by flow cytometry. Immunology 79: 270-277, 1993.
Light scatter analysis, membrane immunophenotyping and determination of DNA content in human whole peripheral blood leukocytes. Cells were stained with anti-CD3 (FL1) and anti-CD8 (FL2), and the fixed with (A) 2% formaldehyde, and (B) solution A.2. Left panels, DLS contour plots; right panels, FL1 and FL2 fluorescences.
Histograms show the 7-AAD fluorescence (FL3-area) of the cells fixed with the A.2 solution shown in panel B: (1) gate R4; (2) gate R5; (3) gate R6.
Figure 2.
Differential ight scatter (DLS) analysis (upper panels) and determination of DNA content (lower panels) in unfixed or fixed rat thymocytes.
(A) Unfixed cells. (B) Cells fixed with the protocol of Telford et al. (Ref. 7). (C) Cells fixed with solution A.2. (D) Cells treated with lysing solution and then fixed with A.2 solution.
Figure 3.
Membrane permeability, cleavage of DNA (TUNEL assay) and DNA content (staining with 7-AAD after fixation with A.2 solution) in rat thymocytes induced to apoptosis by 24-hour in vitro culture.
(A) Light scatters of unfixed cells. (B) Plasma membrane permeability to 7-AAD. (C) Negative control of TUNEL assay (tdt omitted) in cells permeabilised with A.2 and stained with 7-AAD for DNA content. (D) Positive control of TUNEL assay (tdt included) in cells permeabilised with A.2 and stained with 7-AAD for DNA content.
Figure 4.
Rat thymocytes induced to apoptosis by 24-hour in vitro culture.
(A) Light scatters and (B) percent of TUNEL-positive cells (M1, 23%; overlay histogram with the negative control without tdt) in a TUNEL assay performed according to the manufacturer's instructions.
(C) Frequency of hypodiploid cells by the assay of Telford et al. (ref. 7).
(D) DNA staining in cells fixed with solution A.2.
(E) DNA staining in cells treated with lysing solution and then fixed with solution A.2.